在探索物质世界微观奥秘的征途中,科技巨头字节跳动跨界融合创新,成功研发出一种名为ByteFF的高精度通用小分子力场。这一突破性成就标志着人工智能技术与传统化学领域的深度交汇,为药物发现、材料科学等众多领域带来了前所未有的变革潜力。ByteFF不仅提升了模拟计算的精确度,还大大加速了对复杂分子结构行为的理解,降低了研究成本,打开了科学研究的新窗口。通过智能算法的精妙设计,ByteFF能够高效预测小分子间的相互作用,为新药研发提供强有力的支持,预示着个性化医疗和先进材料开发的未来之路将更加宽广而迅速。这一创新之举不仅是技术的跨越,更是对科学探索边界的勇敢拓展。
字节跳动推出全新小分子力场ByteFF:兼顾精度与效率,覆盖广阔化学空间
编辑|ScienceAI
小分子力场在药物发现领域至关重要,是计算机辅助药物设计的核心工具。一个覆盖范围广、精度高的小分子力场,将为药物研发提供坚实基础。
现有基于机器学习的力场(MLFF),例如ANI-2x和MACE-OFF23,虽然能提供精准的分子势能面预测,但却面临数据需求量巨大、计算速度慢以及外推能力不足等挑战,限制了其在药物研发中的实际应用。
Espaloma等一系列研究尝试在保留传统力场势函数形式的基础上,利用图神经网络(GNN)预测传统力场的参数,在精度和效率之间取得平衡,为传统力场的发展指明了新方向。
基于此,字节跳动研究团队开发了ByteFF力场。该力场采用符合物理约束的模型结构,构建了涵盖广泛化学空间的大规模数据集,并设计了相应的训练方案。测试结果表明,ByteFF在结构优化、分子势能面预测等多个方面均达到业界领先水平。
相关研究成果已发表在《ChemicalScience》期刊上,论文标题为“Data-DrivenParametrizationofMolecularMechanicsForceFieldsforExpansiveChemicalSpaceCoverage”。
论文链接:
研究背景
在虚拟筛选、分子对接和自由能预测等计算方法中,小分子力场扮演着关键角色。随着计算机辅助药物设计(CADD)和有机合成技术的进步,药物研发探索的化学空间不断扩大,迫切需要一种能够在广阔化学空间内提供高精度预测的小分子力场。
近年来,量子化学和机器学习技术为传统力场的发展带来了新的机遇。虽然备受关注的MLFF能够提供高精度预测,但其模型复杂性导致训练数据需求量大、计算速度慢等问题,难以兼顾计算效率和化学空间覆盖范围。
2022年,Espaloma力场提出了一种兼顾精度和效率的新方法,即保留传统力场的势函数形式,并使用GNN预测力场参数,取代传统的查表方法。这种数据驱动的参数化方法在提高传统力场精度的同时,保持了计算效率。
在此基础上,提升力场精度和化学空间覆盖率,需要更精巧的模型结构设计和训练策略。
模型结构与训练策略
ByteFF模型结构分为特征提取层(Featurization)、图神经网络层(GNN)和输出层(Output)三部分。
特征提取层将原子和化学键的化学特征转换为向量表示。GNN层采用EGT结构进行信息传递,充分利用原子和键的特征,获得每个原子和键的化学环境表示。输出层根据化学环境预测力场参数。
ByteFF模型结构的设计保证了参数预测结果符合物理约束,例如相同化学环境的结构具有相同的结构参数预测,原子部分电荷之和等于分子的总电荷等。
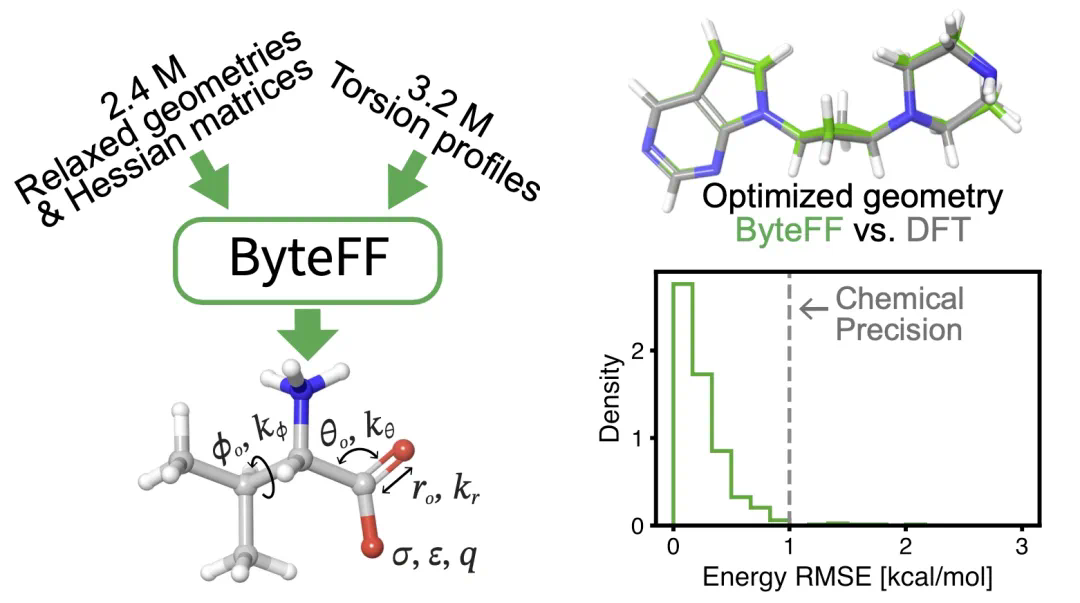
在训练方面,研究团队构建了包含240万个不同分子碎片的优化数据集和320万个不同二面角的扭转数据集。基于这些数据集,研究人员设计了针对性的Hessian损失函数,能够对批量数据进行端到端训练。
由于传统力场形式的限制,难以完美拟合量子化学计算的势能面,研究人员在扭转数据集上采用了迭代的“结构优化-训练”策略,以确保ByteFF在二面角(关键自由度)上提供准确的势能面预测。
此外,研究团队还采用了预训练、训练和微调的多阶段训练流程,以达到最佳训练效果。
性能评估
在结构优化方面,ByteFF显著优于业界领先的OPLS4+ffbuilder(标记为“OPLS4cst”)。

ByteFF能够准确预测小分子(包括环状和非环状分子)的二面角势能面。

更多结果请参考原文。
总结与展望
ByteFF力场凭借其先进的网络结构设计、充足的训练数据和匹配的训练方案,在结构优化和分子势能面预测等方面取得了显著成果。
ByteFF继承了GAFF2的非键参数,保证了与Amber力场的兼容性,但其非键相互作用方面仍有提升空间,这将是未来的研究重点。
目前,ByteFF免费API测试正在进行中,如有需求,请联系论文通讯作者并注明单位和用途。欢迎同行试用并提供反馈。
以上就是AI赋能传统力场:字节跳动开发高精度通用小分子力场ByteFF的详细内容,更多请关注其它相关文章!
 相关攻略
相关攻略 最新攻略
最新攻略












